| المجلد 3 , العدد 5 , شعبان 1425 - تشرين الأول (أكتوبر) 2004 |
| |
| التمييز السيتولوجي والنمط النووي لـ 18 حالة ابيضاض دم سليف نقوي حاد |
Cytological Characterisation and Karyotype in 18 Cases of
Acute Promyelocytic Leukemia |
| Braham-Jmili N.1, Sendi H.2, Omri H.3, Ben Abdelali R.2, Hizem S.1, Laatiri A.3, Saad A.2 and Kortas M.1 |
1- Laboratoire d’Hématologie, 2- Laboratoire de Cytogénétique
3- Service d’Hématologie Clinique
CHU Farhat Hached de Sousse - Tunisia
|
| الملخص Abstract |
المقدمة: يملك ابيضاض الدم سليف النقوي (APL) زميرات (مجموعات فرعية) مُعرَّفة مورفولوجياً ومخصصة بجين سليف نقوي (PML) على الصبغي 15 وبجين مستقبلة حمض الريتونوئيك retinioic ألفا (RAR ألفا) على الصبغي 17.
المرضى والطرق: تمت دراسة 18 مريضاً من عام 1998 إلى عام 2002، 16 أنثى و 5 ذكور بعمر وسيط هو 37 عاماً. اعتمدت المعايير التشخيصية لابيضاض الدم سليف النقوي على تصنيف WHO.
النتائج والمناقشة: أظهرت الدراسة أن التصنيف المورفولوجي بأخذ بعين الاعتبار قَسَمات (ملامع) features هيولية ونووية يمكنها بسهولة تمييز APL عن الأنماط الأخرى للابيضاض النقياني myeloid؛ 16 حالة FABM3 وحالتا تنوع M3 / M3. أظهر النمط النووي karyotype تعبيراً رئيسياً للإزفاء T (15؛ 17) (حاللات 15/ 18)، تبعاً للمعطيات المنشورة (70 – 90%). إن تعيين هذا الإزفاء الصبغي النوعي هام لتأكيد تشخيص APL، للاستجابة المتوقعة لكل من حمض الترانسريتينوئيك وثلاثي أكسيد الأرسينيئك، ولكشف الداء الضئيل المتبقي والنكس relaps المتوقع. |
Introduction: Acute promyelocytic leukaemia (APL) has distinct subgroups defined morphologically and is characterized by the promyelocytic gene (PML) on chromosome 15 and the retinoic acid receptor-alpha (RAR alpha) gene on chromosome 17.
Patients and Methods: 18 patients were studied from 1998 to 2002, 16 female and 5 male with a median age of 37 years. Diagnostic criteria of APL were based on WHO classification.
Results and Discussions: The study revealed that morphologic classification taking into account nuclear and cytoplasmic features can easily distinguish APL from other types of myeloid leukaemia; FAB M3: 16 cases and M3/M3 variant: 2 cases. Karyotype showed mainly expression of translocation t (15; 17) (15/18 cases), in accordance to literature data (70-90%). The determination of this specific chromosomal translocation is important to confirm the diagnosis of APL, to predict response to all transretinoic acid and arsenic trioxide, to detect minimal residual disease and to predict relapse.
|
| Introduction |
|
The world Health organization (WHO), proposed a new classification for hematopoietic and lymphoid neoplasms. Acute leukemia has various clinicopatho-logical and molecular features. A basic principle of the WHO system (2001) is that the classification of acute myeloid leukemia should utilize not only morphologic analyses but also all available information including Immunoph-enotypic, cytogenetic and molecular features for the characte-rization of different types (19).
The purpose of this study is to describe cytologic characteristics of blood and bone marrow at 18 patients with promyelocytic acute leukemia (APL), and to study correlation with karyotype with a review of the recent literature about the different feathers of this disease.
Acute promyelocytic leukemia has distinct subgroups defined morphologically (accounts 10% of all FAB variants). APL is characterized by the promyelocytic gene (PML) on chromosome 15 and the retinoïc acid receptor-alpha (RAR alpha) gene on chromosome 17 (18). |
| Patients and Methods |
|
Patients:
We present 18 patients with acute promyelocytic leukemia; diagnosed from 1998 to 2002 in the area center of Tunisia, 16 females and 5 males. Median age is 37 years.
Methods:
Diagnostic criteria of APL were based as WHO classification (19).
• Cytological investigation
- The hemograms of the 18 patients were done on coulter MAXM blood cell counter.
- Automatic staining of blood and bone marrow smears by using the HEMA TEK slide stainer (AMES company) and a HEMATEK bloc colorant stain pack (Bayer Diagnostica).
- Cytomorphologic exam of the slides for peripheral blood and bone marrow seperatly by 3 morphologists (french-American-British classification (FAB).
• Cytogenetic investigations
Karyotypes were obtained after cultures of the bone marrow cells for 24 and 48 hours, after FrdU-thymidine synchronisation. Metaphase chromosomes were banded by the conventional RHG banding technique. Karyotypes were assigned according to the recommendations of the international System for Human Cytogénétic Nomenclature.
We reviewred the literature on the diagnosis, a cogulopathy, the pathology and current treatment options.
|
| Results and Discussions |
The study revealed that morphologic classification taking into account nuclear and cytoplasmic features could easily distinguish APL from other types of myeloid leukemia
FAB M3: 16 cases
Hypergranular APL/6 (fig 1).
Hypogranular or microgranular variant form (M3v)/10 (fig 2).
M3/M3 variant: 2 cases
Blood: hyperbasophilic form
Bone marrow: M3v.
The initial morphological description of APL emphasized a specific hypergranular, promyelocyte-like, blast cell component in the bone marrow. Although the term promyelocytic leukemia has been widely accepted since 1957 (7, 11) it is partly misleading because APL cells are morpholically different from normal promyelocytes. In 1976, the French-American-British (FAB) cooperative group revised the morphological description of acute myeloid leukemias (AMLs); the term M3-AML was assigned to hypergranular promyelocytic leukemia characterized by blast cells with heavy azurophilic granules, bundles of Auer rods (faggots), and a reniform or bilobed nucleus (11).
Major interest has focused on APL following the identification of a specific cytogenetic abnormality: t (15; 17) (q22; q21) translocation (17). Although the vast majority of M3 cases fit the description of hypergranular or classical M3, a cytological hypogranular or microgranular variant form, M3v, has been identified. It is commonly associated with hyperleukocytosis (10), accounts for 15% - 20% of APL cases, and shares the same t (15; 17). In M3v-AML, the majority of blasts have a bilobed, multilobed, or reniform nucleus and, under usual staining, are devoid of granules or contain only a few fine azurophilic granules; however, at least a few cells with all the cytoplasmic features of M3 are present (3). Rare morphological forms associated with t (15; 17) have been reported. These cases are characterized by hyperbasophilic
microgranular blast cells with cytoplasmic budding that mimicks micromegakaryocytes (2, 14) or by blast cells that exhibit toluidine blue positive basophilic granules (5, 12) or by eosinophilic granules. 19 other cases have been reported to have M1- or M2- like morphology (1, 15).
|
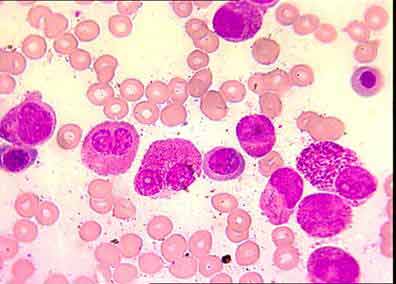
Fig. 1: hypergranular cytoplasm, 3 or more Auer rods,
reniform nucleus, irregular nucleus
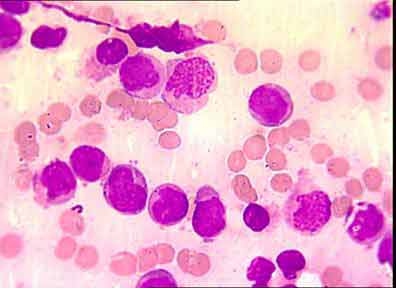
Fig. 2: hypergranular or agranular cytoplasm, irregular nucleus.
|
Table I : Results of cytological and cytogenetic investigations for the 18 patients with acute promyelocytic leukemia.
Analyse |
Number of cases |
Myelograms |
Diagnosis of APL (1) : 18 cases |
Hemograms |
Pancytopenia: 14 cases
Leukocytosis (WBC (2) >50 000/mm 3 ): 4 cases |
Karyotypes |
t (15;17): 15 cases |
(1): APL: Acute Promyelocytic Leukemia.
(2): WBC: White Blood Cell.
|
In 1990, t (15; 17) was cloned and was found to disrupt a previously uncharacterized gene, PML, on chromosome 15 and the gene encoding the retinoic acid receptor alpha (RARA) on chromosome 17. The resultant PML/RARA fusion protein, which retains the retinoic acid (RA) ligand-binding domain, not only plays a key role in leukemogenesis, but it is also somewhat paradoxically implicated in mediating the response to retinoids (6, 8).
The karyotype revealed the expression of translocation t (15; 17) in 15/18 case (Table I), in accordance to literature data (70-90%). In deed, approximately 10% of all APL lack the classic t (15; 17). This group includes (18):
- Cases with cryptic PML/RARA gene rearrangement and t (5; 17) that leads to the NPM/RARA fusion gene which are retinoid responsive.
- Cases with t (11; 17) (q23, q21) that are associated with the PLZF/RARA fusion gene which are retinoid resistant.
Our results show that there were no major differences observed in morphology between cases with classic t (15; 17) and normal karyotype.
APL provides a useful model in witch therapy is targeted to an underlying genetic aberration and treatment is adapted, based on monitoring of residual disease (6, 8)
APL was first recognized for its particularly poor clinical out come due to a serious hemorrhagic syndrome, which is related to disseminated intra vascular coagulopathy and abnormal fibrinolysis (18).
Discarded homeostasis is typical of acute promyelocytic leukemia (FAB M3) and relates to the intrinsic properties of the blast cells as well as thrombocytopenia from bone marrow involvement (13).
APL promyelocytic leukemia has become the most potentially curable subtype of AML in adults because of the introduction of novel approaches in the management of this disease (8, 9). All transretinoïc acid (ATRA) based therapy is now the first choice treatment of patients presenting with de novo APL; based on induction therapy followed by two or three courses of consolidation chemotherapy and clinical studies have shown that currently around 90% of newly diagnosed patients who receive ATRA therapy achieve complete remission and over 70% of patients are curable (4). Arsenic trioxide (ATO) has been established as highly effective therapy for patients with APL (16).
|
| Conclusion |
|
This study highlights:
-The importance of the determination of the specific chromosomal translocation t (15, 17): to confirm the diagnosis of APL, to predict a beneficial response to all transretinoïc acid and arsenic trioxide, to detect minimal residual disease and to predict relapse
-The importance of combining morphologic and cytogenetic analyses for optimal management of APL patients and better understanding of pathogenesis of the disease. |
| References |
1-Allford S; Grimwad D; Langabeer S. et al.
On behalf of the Medical Research Council (MRC) adult leukemia working party. Identification of the t (15; 17) in AML FAB types other than M3: evaluation of the role of molecular screening for the PML/RAR alpha rearrangement in newly diagnosed AML.
Br J Hematol, 105: 198-207, 1999.
2-Aventin A; Maten R; Martino R; Colomer D. and Bordes R.
A case of cryptic acute promyelocytic leukemia.
Leukemia, 12: 1490-1506, 1998.
3-Benett J.M; Catovsky D; Daniel M.T. et al.
A variant form of acute hypergranular promyelocytic leukemia (M3).
Br J Haematol, 44: 169-170, 1980.
4-Callager R.E.
Retinoïc acid resistance in acute promyelocytic leukemia.
Leukemia, 16: 1940-1958, 2002.
5-Castoldi G.L; Liso V; Specchia G. and Tomasi P.
Acute promyelocytic leukemia: morphological aspects.
Leukemia, 8: 1441-1446,1994.
6-Coco F.L; Diverco D; Folini B; Biordi A; Nervi C. and Pelicci P.G.
Genetic diagnosis and molecular monitoring in the management of acute promyelocytic leukemia.
Blood, 94: 12-22, 1999.
7-Degos L.
The history of acute promyelocytic leukemia.
Br J Hematol, 122: 539-553, 2003.
8-Dover D.
New advances in the treatment of acute promyelocytic leukemia.
International Journal of Hematology, 76 (2): 179-187, 2002.
9-Fatanga A. and Barbui T.
Coagulopathy of acute promyelocytic leukemia.
Acta Hematologica ,106: 43-51, 2001.
10-Golomb H.M; Rowley J; Vardiman J.W; Testa J.R. and Butler A.
“Microgranular” acute promyelocytic leukemia a distinct clinical, ultra structural and cytogenetic entity.
Blood, 55: 253-259, 1980.
11-Hillestad L.K.
Acute promyelocytic leukemia.
Acta Med Scand, 159: 189-195, 1957.
12-Invernizzi R; Lannone A.M; Bernuzzi S. et al
Acute promyelocytic leukemia toluidine blue subtype.
Leuk Lymphoma, 18: 57-60, 1995.
13-Kalke E; Goede A. and Rose P.
Acute arterial thrombosis in acute promyelocytic leukemia.
Clinl Lab Hematol, 25 (4): 267-270, 2003.
14-Kenna R.W; Parkin J; Bloom field C.D; sundberg R.D. and Brunning R.D.
Acute promyelocytic leukemia: a study of 39 cases with identification of a hyperbasophilic microgranular variant.
Br J Haematol 44: 169-170, 1982.
15-Neame P.B; Soamboonsrup P; Leber B. et al
Morphology of acute promyelocytic leukemia with cytogenetic or molecular evidence for the diagnosis: characterization of additional microgranular variants
Am J Hematol 56: 131-142, 1997.
16-Ohno R; Asou N. and Ohnishi K.
Treatment of acute promyelocytic leukemia: strategy toward further increase of cure rate.
Leukemia, 17 (8): 1454-1463, 2003.
17-Rowley J.D; Golomb H.M. and Dougherty C.
15/17 translocation, a consistent chromosomal change in acute promyelocytic leukemia.
Lancet, 1: 549-550, 1977.
18-Stainy D; Liso V; Rajnoldi A. C. et al.
A new morphologic classification system for acute promyelocytic leukemia distinguishes cases with underlying PLZF/RARA gene rearrangements.
Blood, 96(4): 1287-1296, 2000.
19-Vardiman J.W; Harris N.L. and Brunning D.
The world health organization (WHO) classification of the myeloid néoplasms. Blood, 100 (7): 2292-2302, 2002.
|
| |
| المجلد 3 , العدد 5 , شعبان 1425 - تشرين الأول (أكتوبر) 2004 |
|
|
|
