| المجلد 3 , العدد 7 , صفر 1426 - نيسان (أبريل) 2005 |
| |
| مراجعة في الوراثة اللاجينية، وارتباطها بالوراثة |
| A Review in Epigenetic, and its Linking to Heritance
|
| د. رسلان العلي و د. سحر الفاهوم (مقال مرجعي باللغة الإنجليزية) |
| Alali R. and Al-Fahoum Sahar
|
| كلية الصيدلة، جامعة دمشق |
| Abstract |
| إنها حركة جديدة إلى الأمام في العالم العلمي للجينات. تلك العديلات اللاجينية التي يتم التعامل معها في هذه المراجعة والتي تتضمن أستلة، وفسفتة ومثيلة الهسيتونات، ومثيلة الدنا DNA وإعادة طراز الكروماتين المعتمدة على الـ ATP. وإنه منظر يلمح إلى التغذية وبعض الحالات الهامة التي تفسر التعديلات بالطريق اللاجينيي مثل تعطيل الصبغي X والخروف الكاليبيج Callipyge sheep. |
| A new headway of the scientific world of genes. Epigenetic modifications that this review deals with include acetylation, phosphorylation and methylation of histones, methylation of DNA, and ATP-dependant chromatin remodeling. A glance view of nutrition and some important cases that is explained via epigenetic modifications like chromosome X inactivation and callipyge sheep. |
| Introduction |
A few decades ago, two scientists discovered a double stranded helix of deoxyribose nucleotides we know it now simply as DNA. DNA was proved, long time ago, to be the molecule responsible for inheritance of apparent characteristics and diseases in living organisms. DNA is considered as the blueprint of all proteins in the cell, any protein production should start from DNA.
Tow groups of scientist sequenced human DNA, the final draft was revealed first at 3/2004, a database of more than 3 billion bases of cytosine (C), guanine (G), thiamine (T), and adenine (A). It was just the beginning not the end as everyone thought.
The genome is the data stored in chromosomes and that control the way the organism grow. It is thought to be a 3D machine that transfers data to the next generation.
The coding genes of DNA constitutes about 2% of whole DNA in each human cell, leaving the rest 98% as a junk material. While blaming the "active" DNA for all of the genetical diseases. As theory goes that only genes, pieces of DNA that finally produces proteins, are the parts that we need to care about.
In early sixties, it was pointed to hidden places in non-coding DNA as being responsible for many hereditary incidents (see chromosome X, solid gold later). Such a note didn't bring much attention of the scientific society to study the "long forgotten 98%" till recently.
Countless RNA-only genes were found. These missing parts play a significant role in controlling protein expression. And any mutations in those genes can lead to a hazard complications.
The protein is another story, DNA is not the only hereditary material, proteins form most of the chromosomes and they affect the statues of gene expression. They can accumulate and make DNA unreachable to transcription enzymes thus make the gene silent.
DNA forms a doubles stranded helix that turns around a ball-like complex of proteins, these proteins are known as histones, there are five types of histones: H2A, H2B, H3 and H4 (two molecules for each histone-ball) and one histone H1. They are positively charged at neutral pH, they contain 20% lysine and arginine of their amino acid structure. Most of them are conserved from one organism to another which dictates a hypothesis that they have principal participation in DNA's appropriate function. Folding of nucleosomes* forms a solenoid model, which forms loops and super-loops later till it reaches the final shape of chromosome. The looping is not uniformed, the chromatin** can be dense in some places and loose in others. Many mechanisms control these variations. In addition, there are gaps in the series of nucleosomes so that nucleosomes-free DNA regions is detected (2, 14). Once chromatin condensation starts, it continues along the chromosome till it reaches such a gap (1).
Chromatin can depress gene work when it is dense and activates its work when the chromatin is relaxed. Epigenetic changes? include acetylation, phospho-rylation and methylation of histones which affect the tightness of DNA turns around the histones, leading to a change in the ability to express the gene by preventing transcription by RNA-polymerases due to the attachment of several proteins (15). Methylation of DNA itself and ATP-chromatin remodeling can be added to these changes. |
*Nucleosome: A repeated structural element in eukaryotic chromosomes, composed of core of eight histone molecules with about 200bp of DNA wrapped around the outside and one molecule of histone H1 also bound outside the core histone octamer.
**Chromatin: The material of chromosomes, composed of DNA and chromosomal proteins.
?Epigenetic change: A change in the expression of a gene, not in the structure of gene itself.
It started with acetylation
In 1964, Allfrey discovered that histones have two forms; acetylated and unacetylated. Acetylation occurs on amino groups of lysine and arginine side chains. Allfrey showed that acetylation of histones correlates with gene activity, while deacetylated histones tend to repress transcription. These findings implied that enzymes in nuclei acetylate and deacetylate histones and thereby influence gene activity. Figure 3.
In 1996, Brownell and Allis purified a histone acetyltransferase (HAT), an enzyme that transfers acetyl groups from acetyl-CoA to core histones (2). This enzyme adds the acetyl group to ? amino group which is often the evolutionary conservative lysine residues most often located in tail domains of histones (3, 11, 12). Fully acetylated H3 has acetyl group on lysines 9, 14 and 18 and fully acetylated H4 has acetyl group on 5, 8, 12 and 16. Lysines 9
and 14 of histones H3 and lysines 5,8 and 16 of histone H4 are acetylated in active chromatin and deacetylated in inactive chromatin (2), Figure 2.
|
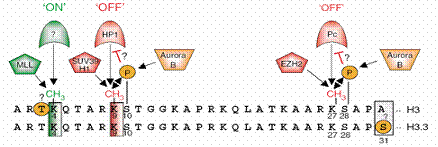 |
| Figure 1: Amino acid sequence in histone H3 and one of his variants: H3. 3, and the point which epigenetic actions take place at, with proteins that associate with it afterwards. Pentagon shapes at the left side of each site is the enzymes that affect this site. Dominant methyl marks are found on lysines 4, 9, and 27 in histone H3, all adjacent to threonine or serine, potential phosphomark carriers. Methylation of lysine 4 is generated by the SET domain of MLL, and is connected to gene activation of certain target genes (green¼‘ON’ mark). Protein(s) that ‘read’ this mark are not yet identified. Marks that correlate with gene silencing are methylation of lysines 9 and 27, generated by the HMTs SUV39H1 and EZH2, respectively (red¼‘OFF’ marks). ‘Readers’ of these repressive marks are HP1 for lysine 9 methylation and Pc for lysine 27 methylation. Serines, adjacent to lysines 9 and 27, are shown to be phosphorylated by Aurora B kinase (orange), and might play a role in preventing ‘readers’ from recognizing methyl marks. It is not yet known if threonine 3 is also a phospho mark (orange circle). Sequence alignment of the N-termini of H3 with H3.3 variant shows an almost identical sequence, except that alanine 31 in H3 is replaced by serine in H3.3, another potential phospho mark (3). |
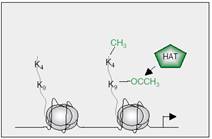 |
| Figure 2: Histone acetyltransferase (HAT) adds and acetyl group to lysine 9 on the tail of histone H3 making the gene active. A methyl group can be added on lysine 4 too (see methylation later) (3).
|
| While the process of deacetylation is completed by histone deacetylases (HDACs) which not only leads to chromatin compression but it also make the nucleosomes more attached to each other (2), hence leads them to be inactive (or at least less active). |
| Phosphorylation also matters |
Phosphorylation is another important and long-appreciated histone modification that is often associated with chromosome / chromatin condensation that includes mitosis, meiosis, apoptosis and DNA damage, events regulated by different histone kinases (for example, members of the Aurora/AIK family)(19). Histone phosphorylation is also closely correlated with chromosome decondensation events (such as the immediate-early response to mitogens), suggesting a ‘split personality’ for certain modifications that remains to be fully understood (for example, at serine 10 in H3) (3, 18), Figure 1.
Also, phosphorylation can be one of the regulatory factors in ovarian function, it's due to its correlation with the effects of gonadotropic hormones from pituitary gland and estrogens from the ovary. Immuno-histochemical evidence indicates
that histone H3 phosphorylated on Ser10 was found in punctuate distribution (associated with gene expression) and a condensed distribution in granulosa cells. FSH and estrogen induce phosphorylation of histone H3 on Ser10 and its acetylation on Lys9 and Lys14 in rat granulosa cells, each one of these hormones has its phosphorylation effect independently of the other. Knockout mice for estrogen receptor-? had an impaired H3 phosphorylation while knockout for estrogen receptor-? didn't affect this process (9).
Phosphorylation is like the acetylation, it is a short term epigenetic factor.
|
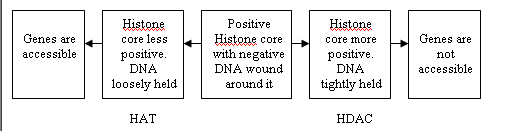
Figure 3: Function of histone acetyltransferase (HAT) and histone deacetylase (HDAC). The effect of adding an acetyl with HAT in activating genes, and inactivating it via HDAC when an acetyl is removed from histones' tail. |
"Methyl" is the password
The most recognized change in both histones and DNA is methylation.
Starting with one carbon and three hydrogen atoms, this group strongly tends to stick on any thing, the reaction is called methylation.
Like in acetylation, the methylation of histones play a major role in controlling gene expression, but it has a different method of action. Methylation of histones occurs on lysine and arginine residues. Lysine residues is a well known target as an acetylation sites too (see acetylation previously mentioned),
for example lysine 9 in histone H3. Mono-, di- or tri-methylated lysine and mono- or di-methylated arginine can complex the modification-dependent gene regulation. The dynamic arginine methylation in H3 and H4 is correlated with gene activation, and its loss from target arginines correlates with gene inactivation.
While the methylation of lysines appears to be more stable and more complicated. For example, as depicted in Figure1, methylation of lysine 4 in H3 is correlated with gene inactivation, whereas methy-lation of lysine 9 and 27 in H3 correlates with repression. The methyl groups is attached by histone methyltransferases (HMTs) which contains a SET (Su (var) 3-9, Enhancer-of-zeste, Trithorax) domain.
|
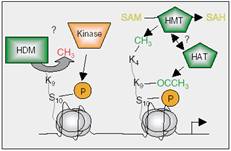 |
| Figure 4: Although there is no HDM identified to date, one canspeculate that, if this enzyme exists, serine 10 phosphorylation in H3, for example, by Aurora kinases, can lead to recruitment of HDMs that in turn demethylate lysine 9 in H3. Histone acetyltransferases might then acetylate lysine 9 and HMTs methylate lysine 4, resulting in the loosening of the chromatin structure and allowing gene transcription.
|
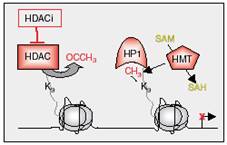 |
| Figure 5: Gene repression through histone methylation. Histone deacetylase deacetylates lysine 9 in H3, which can then be methylated by HMTs. Methylated lysine 9 in H3 is recognized by HP1, resulting in maintenance of gene silencing (3). |
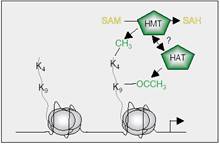 |
Figure 6: Setting ‘ON’ marks in histone H3 to activate gene transcription. Lysine 4 in H3 is methylated by HMT (for example MLL) and lysine 9 is acetylated by HAT, allowing genes to be transcribed. It is not known, if HMTs and HATs have a direct connection to each other (3).
Methylation on lysine 4 of H3 inhibits binding of chromatin-remodeling NuRD (see chromatin remodeling later) and HADCs that deacetylates histone tails (see acetylation above). In addition, this blocks methylation of nearby lysine 9 and acetylates this site, Figure 6, the two effects mentioned above maybe the cause of activating the gene with methylated-lysine 4 of its H3 (2, 3).
When lysine 9 is methylated via methyltransferase (SUV39H) it is vulnerable to the attack of a histone methyltransferase associated protein that's called HP1, Figure 1. HP1 could then recruit SUV39H (the HMTase) to methylate another nearby histone H3 on its lysine 9. (2, 15). In this way the process continues for several nucleosomes and usually stops at the gaps of nucleosome-free DNA. The methylation of this lysine can be inhibited by several modifications on the same histone tail, including acetylation of lysine 9 (and perhaps lysine14), methylation of lysine 4 and phosphorylation of serine 10, figur 4. (2).
On the other hand, in DNA itself the most favorite target to be reacted with is Cytosine group (C). One of the most characteristic methylated sites on DNA is CpG (adjacent cytosine and guanine nucleotides) islands (1, 3, 7). Hyper-methylation of those islands is associated with transcriptional silencing of the genes which CpG islands have transcriptional control upon, as this sites are considered as a famous promoter (2, 4).
The enzymes responsible for that are the family of DNA methyltransferase family (DNMT) which catalyze the addition of methyl group to cytosine residues at CpG islands, by converting SAM to SAH, Figure 7. A family of proteins with methyl-binding domain (MBD) can recognize methylated DNA, Figure 8, and have been shown to associate with large protein complexes containing HDACs and chromatin-remodeling activities. As a result, histones are deacetylated and gene transcription is most often repressed. It has also been suggested that DNA methylation could lead to gene
silencing by MBD proteins that recruit HMTs which methylate lysine 9 in histone H3 and subsequently repress gene transcription (3).
|
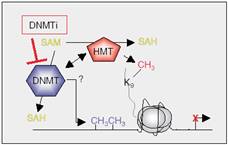 |
| Figure 7: Interplay between DNMTs and HMTs results in methylation of DNA and lysine 9 in H3, and consequent local heterochromatin formation. The exact mechanism of this cooperation is still poorly understood. |
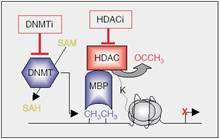 |
| Figure 8: Gene repression involving DNA methylation. DNA methyltransferases methylate DNA by converting SAM to SAH, a mechanism that can be inhibited by DNMT inhibitors (DNMTi). MBPs recognise methylated DNA and recruit HDACs, which deacetylate lysines in the histone tails, leading to a repressive state. |
Methylation imprint the DNA, but it is removed in the primordial germ cells (PGCs). This appears to be an active demethylation process, involving yet-unknown enzymatic activities. Novel imprints are established much later during gametogenesis, at late foetal stages in male germ cells, or after birth in growing oocytes (31). This is the reason why two twins having the same DNA may differ in genetic characters, same applies on clones (1).
Chromatin remodeling
Methylation, acetylation and phosphory-lation are not the only changes that occur to the chromatin. Acetylating core histone tails can disrupt nucleosomes cross-linking but it leaves nucleosomes intact. So chromatin needs remodeling to allow transcription factors to access, this activity requires ATP. There are the SWI/SNF family, the ISWI family, the NuRD family and the INO80 family. All four classes of proteins alter the structure of nucleosomes cores to make DNA more accessible (2, 3, 34). All these families may yield the nucleosome-free regions around enhancers and promoters that are characteristic of active genes (2, 34).
|
Diet and epigentics
Agouti mice hair-color ranges between yellow and black. The expression of yellow hair color is controlled by hypomethylation of agouti mice long terminal repeats (LTR). When normal maternal diet was supplied, more than 60% of their offspring were yellow mice. But when they were fed nutrients rich with vitamin B12, folic acid, zinc, methionine, betaine and choline, the expression in 60% of their offspring were agouti gray. Such dietary change may caused hyper-methylation of LTR and accordingly a change in the color (1, 21).
S-adenosylmethionine (SAM) and S-adenosylhomocysteine (SAH), as components of methyl metabolism, are the substrate and product of essential cellular methyltransferase reactions. SAM is derived from an ATP-dependent transfer of adenosine to methionine via methionine adenosyltrans-ferase and serves as the proximal methyl donor for most methylation reactions. Cellular methyl acceptors include phospholipids, proteins, histones, neurotransmitters, RNA and DNA. The formation of SAM requires a continuous supply of folate, methionine, choline, vitamin B12, vitamin B6, vitamin B2 and energy from the extracellular milieu (21).
Stress including that resulting from dietary methionine/choline, folate, zinc, and selenium inadequacy, as well as excessive alcohol intake can lead to global DNA hypomethyl-ation. Global hypomethylation, accompanied by region-specific hypermethylation, is a common characteristic among tumor cells. Global genomic hypomethylation has been linked to the induction of chromosomal instability. Hypermethylation is associated with the inactivation of virtually all pathways involved with the cancer process, including DNA repair (e.g., hMLH1, BRCA1, MGMT), cell cycle regulation (e.g., p16 (INK4a), PTEN), inflammatory /stress response (e.g., COX-2) and apoptosis (e.g., DAPK, APAF-1) (21, 35).
|
RNA that stops the DNA
RNA molecule is known to have three forms: mRNA, tRNA and rRNA. But there are many cases where RNA reported to be an active form that controls the expression of DNA, either by inactivating mRNA via making it vulnerable to RNases and called RNA interfe-rence (RNAi), or by affecting the DNA itself causing inactivation of the gene, several genes or even the whole chromosome (see cases Callipyge ram and chromosome X inactivation later).
RNAi is considered a very promising technique that has been used to inactivate the expression of genes and been studied in order to use in cancer therapies. While the second type of RNA works when attached to a specific site of DNA and initiates its methylation, Figure 9. The last form of RNA is produced from RNA-only genes, with a possibility of producing it from dsRNAi (31).
Callipyge ram (1, 24)
A ram were produced in a sheep breeder in Oklahoma, it was called "solid gold". The scientist called the new species: "Callipyge" (i.e. Beautiful Buttocks). It had a big and fat gluteus. Its offspring didn't follow the traditional inheritance laws, Figure 10. Half of the first generation had big gluteus, which is normal for dominant gene expression. But the second gene-ration didn't present the big gluteus when the mother had the mutation. Further more the homozygote for this mutation didn't express this characteristic either (see question marks in Figure 10).
|
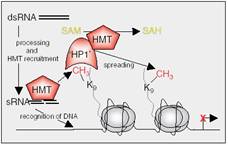 |
| Figure 9: Specific gene repression by small RNAs (sRNAs). Transcription of repetitive DNA sequences lead to double-strand RNA (dsRNA) generation by still poorly understood mechanisms, and dsRNA is later processed to sRNAs. sRNAs associate with and recruit HMTs to the complementary DNA sequence, where HMTs locally methylate lysine 9 in H3. Methylated lysine 9 is recognised by ‘HP1’ that forms a complex with HMTs to spread the repressive mark to other histones, until reaching a boundary. ‘Writers’ and ‘readers’ of DNA epigenetic marks are shown in blue, and proteins involved with repressive histone marks are depicted in red. Tail length has been exaggerated for clarity (3). |
The explanation includes that a mutation, Table 1, from A to G in some unknown-gene of DNA, deactivates some RNA-only genes on the same chromosome. RNA-only genes stop the production of one protein (or active RNA).
The maternal allele is silenced by
epigenetical effects and don't produce mRNA. That's why the maternal gene didn't work to produce proteins. And couldn't stop the production of RNA-only genes. The later thereby stops genes that produce the protein in the paternal chromosome.
|
Table 1: factors that affect "callipyge mutation".
Name of factor |
work |
Gene of protein |
Production of protein or (active RNA), Inhibited by RNA-only genes |
RNA-only genes |
Regulates protein production, inhibited by mutation(A to G). active in both maternal and paternal chromosomes. |
Mutation A to G |
Controls RNA-only genes on the same chromosome , so increase protein production. Controlled by epigenetic maternal-gene modifications. |
Maternal-gene modification |
inhibits Maternal "mutation A to G" so RNA-only genes is activated on maternal chromosome so they inhibit paternal gene of protein, and protein is not produced. |
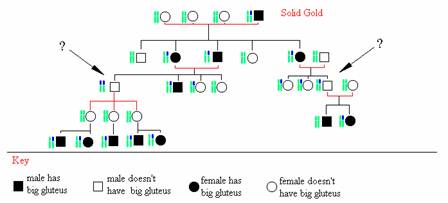
Figure 10: the inheritance of callipyge mutation (blue chromosome) is not submitted to normal lows. The homozygote don't show the big gluteus, and so for all the sheep which inherited the mutation for the mother side. |
Choromosome X inactivation
Two copies of chromosome X are available in female cells, yet only one of them is active, the other is almost completely inactivated.
A gene called Xist produces active RNA that encapsulate the undesired chromosome totally (1, 35), while the other chromosome X produces antisense-RNA (Tsix) to protect itself of Xist gene.(1, 35) The Xist gene is expressed exclusively from the inactive X (7).
On the inactive chromosome, we can notice three major changes: A lot of methyl groups on the DNA, releasing of acetyl groups from histones' tails and chromatin compression to form RNA-closed mass that cannot be penetrated (1).
|
Cancer from epigentical view
The major step in gene regulating is gene inactivation by hyper-methylation of CpG islands located on promoter region. One approach to cure cancer is to inhibit this inactivation of tumor suppressor genes using a chemical agents such as 5-azacytidine to prevent hypermethylation of DNA. (24)
General hypomethylation is associated with local hypermethylation of CpG island so it can silence a tumor suppressor gene by hypermethylation, loss of imprinting and ,less likely activating oncogenes by demethylation. Genetic changes include point mutations at methylated CpG islands that lead to hypermethylation and microsatellite instability owing to silence of repair genes (e.g. MLH1)(35).
Other scientists just care about the methylating factors and their correlations with epidemiology.
Investigators are currently exploring the interaction between these polymorphisms, diet and DNA methylation. For example, subjects homozygous for the C677T polymorphism in the MTHFR gene exhibited a significantly lower level of methylated DNA but only under conditions of low folate status. Poly-morphisms in the methyl metabolism genes methionine synthase, methylenetetrahydro-folate reductase (MTHFR), and cystathione beta-synthetase affect concentrations of SAM and likely modify the susceptibility to cancer.(21, 26) While some reports indicated that folate supplement may lead to cancer condition, such as breast cancer, others disagree. As in a follow up study in Abereen and Bristol it was found that were that were given a higher supplement of folate during early pregnancy had 20% higher mortality risks and two times greater risk of breast cancer (29). But in a study of NCI, women with high folate levels (greater than 14ng/mL) in plasma had a 27% lower risk of breast cancer than did women with a lower level (6.4ng/mL). The high vitamin B6 levels (greater than 95.3 pmol/mL) were associated with a 30% risk reduction as compared to low levels(less than 28.5 pmol/mL). Vitamin B12 levels made a difference in premenopausal women (lower risk) but not among post menopausal women (30).
Experiments on rat showed a miscorrelation with low folate intake and an decrease in MNU-induced tumorigenesis comparing with normal and high folate intake. No significant change were detected in DNA methylation, it was suggested that dietary folate deficiency of moderate degree suppresses mammary tumorigenesis (25).
In addition, experiments showed that increased yellow color in the agouti mice strain (previously mentioned) is linked with increased risk of obesity, diabetes and cancer (21).
In cancer, there is compelling evidence that epigenetic marks, such as chromatin modification, can influence phenotypes through the regulation of particular genes, without an underlying sequence variant in the gene. Alterations in methylation, imprinting and chromatin are ubiquitous in cancer. A causal role is suggested by recent data showing a gatekeeper role for altered imprinting and methylation in Wilms’ tumor, and a common epigenetic variant appears to be associated with risk of colorectal cancer.
|
Detect an epigenetical change
An epigenetical approach could explain many puzzling cases of cancer, it makes the "big image" even harder to see. But new tests to the epigenetic modification using chromatin immunoprecipitation (ChIP) techniques for example, can help scientists to do their job in a better way. ChIP is based on an immuo-test using antibodies towards targeted methylated genes, it can precipitate the methylated strands and separate them easily by centrifuge. Using PCR technique to amplify the portion of interest can detect whether the DNA is bond to proteins or not.
Staining the desired region with labeled antibodies, and using RNase T1 cleavage and MALDI-TOF can be applied too (4).
The most recommended way is using ChIP reaction with microarray (27, 28).
Conclusion
Epigenetic changes take place in human cell since its early beginnings. Acetylation and phosphorylation take place on histone tails and mark the gene for a short period of time, then special enzymes remove it. On the other hand, methylation is a long-term change happens to both histones and DNA. In addition to the changes of chromatin itself, the function of DNA is highly related to these changes. New ways is present, or will be available in near future, to deal with unexplained inherited changes and malfunctions.
Diseases such as cancer will have a new approach to deal with. And cases like callipyge ram will help in providing a better understanding to epigenetic, and will means much more effective way to get the most benefit out of it.
|
References
1-W.W. Keps
Translated by S. Al-Fahoum et al. The unseen genome: Beyond DNA.
Scientific American (Arabic version). 20, 2/3, 2004.
2-R. Weaver
Chp13, Chromatin Structure and Its Effects on Transcription. from Molecular Biology, 3rd edition. 2005. McGRAW HILL
3-M. Esteller
Cancer epigenetics: DNA methylation and chromatin alteration in human cancer.
From publications of Cancer Epigenetic Laboratory. Spanish National Cancer Center (CNIO).
4-Phillip Schatz, Dimo Dietrich and Matthias Schuster
Rapid analysis of CpG methylation patterns using RNase T1 cleavage and MALDI-TOF. Nucleic Acids Research. 32, 21. Oxford University Press 2004.
5-Jonathan M. Levenson and J. David Sweatt
Epigenetic Mechanisms In Memory Formation.
Nature Reviews Neuroscience, 6, 108-118. 2005.
6-Yamasaki H; Omori Y; Daidan-Dagli M-L; Mironov N; Mesnil M. and Krutovskikh V.
Genetic and Epigenetic Changes of Intracellular Communication Genes During Multistage Carcinogenesis.
Cancer Detection and Prevention, 23(4), 273-279, 1999,
7-Allaman-Pillet N; Djemai A; Bonny C. and Schorderet DF.
Methylation status of CpG sites and methyl-CpG binding proteins are involved in the promoter regulation of the mouse Xist gene.
PMID: 9699479
8-Lewin J; Schmitt A; Adorj?n P; Hildmann T. and Piepenbrock C.
Quantitative DNA methylation analysis based on four-dye trace data from direct sequencing of PCR amplificates.
Bioinformatics, Oxford University Press, 2004.
9-Murphy B.
Epigenetic factors regulating ovarian function. Publications of University of Montréal.
10-Kenji Yasuda.
Biotechnology approach to determination of genetic and
epigenetic control in cells.
Journal of Nanobiotechnology, 22 November 2004
11-Strahl B.D. and Allis C.D.
The language of covalent histone modifications.
Nature, 403: 41-45, 2000.
12-Turner B.M.
Histone acetylation and an epigenetic code.
Bio-Essays, 22: 836-845, 2000.
13-Yong-hui Jiang, Jan Bressler, and Arthur L. Beaudet
Epigenetics and Human Disease.
Annual Review of Genomics and Human Genetics, 5: 479-510, 2004.
14-Grewal SI. and Moazed D.
Heterochromatin and epigenetic control of gene expression.
Science, 301: 798 –802. 2003.
15-Jenuwein T. and Allis CD.
Translating the histone code.
Science, 293: 1074-1080, 2001.
16-Mizuguchi G; Shen X; Landry J; Wu WH; Sen S; and Wu C.
ATP-driven exchange of histone HZAZ variant catalyzed by SWR1 chromatin remodeling complex.
Science, 16: 343-348, 2003.
17-Venkat C.
Genes Sleeping on the Job.
Epigentics, November 26, 2004.
18-Cheung P; Tanner KG; Cheung WL; Sassone-Corsi P; Denu JM. and Allis CD.
Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation.
Mol. Cell, 5: 905-915, 2000.
19-Fischle W; Wang J. and Allis CD.
Binary switches and modification cassettes – novel concepts in histone biology and beyond.
Nature, 425: 475-479, 2003.
20-Epigenetics
Novartis institutes for biomedical research publications.
21-Diet, DNA Methylation and Other Epigenetic Events, and Cancer Prevention:Competing Supplements.
National Cancer Institute (NCI). Release Date: September 27, 2002.
22-Jones PA. and Laird PW.
Cancer epigenetics comes of age.
Nat. Genet. 21(2): 163-167, 1999.
23-Jain PK.
Epigenetics: the role of methylation in the mechanism of action of tumor suppressor genes. Ann. N. Y. Acad. Sci., 983: 71-83, 2003.
24-Edward R. Winstead.
Comparing human and sheep genomic sequences six imprinted genes at the callipyge locus.
Genomic News Network. 7, 2001.
25-Kotsopoulos J., Sohan KJ, Martin R., Choi M., Renlund R., Mckerlie C., Hwang S.W., Medline A. and Kim Y.I.
Dietary folate deficiency suppresses N-methyl-N-nitrosourea-induced mammary tumorigenesis in rats.
Carcinogenesis, 24(5): 937-944, 2003.
26- Slattery M., Potter J., Samowitz W., Schaffer D. and Leppert M. Methylenetetrahydrofolate Reductase, Diet, and Risk of Colon Cancer.
Cancer Epidemiology, Biomarkers & Prevention, 8, 513-518, 1999.
27-The website of Roche for diagnostic.
28-The web site of Epigenomics diagnostics.
29- Ness A. and Oaklay G.
Folate supplement during pregnancy may increase a mother's long-term risk of breast cancer.
News-medical, 13 Dec. 2004.
30-Zhang SM, et al.
Plasma folate, vitamin B6, homocysteine and risk of breast cancer.
Journal of the National Cancer Institute, 95(5): 373-380, 2003.
31-Katia Delaval and Robert Feil.
Epigenetic regulation of mammalian genomic imprinting.
Current Opinion in Genetics & Development, 14, 188-195, 2004.
32-Hans T; Bjornsson M; Daniele Fallin and Andrew P. Feinberg.
An integrated epigenetic and genetic approach to common human disease.
Trends in Genetics, 20, 8, 2004.
33-Peters J A.; Mermoud D; O'Carroll Pagani M; Schweizer D; Brockdroff N. and Jenuwein Th.
Histone H3 lysine methylation is an epigenetic imprint of facultative heterochromatin. Website of
34-Michael D. Blower and Gary H. Karpen.
The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions.
Nature Cell Biology, 3, 2001.
35-Rudolf Jaenisch and Adrian Bird.
Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals.
Natural genetic supplement, 33, 2003.
|
| |
| المجلد 3 , العدد 7 , صفر 1426 - نيسان (أبريل) 2005 |
|
|
|
