| المجلد 5 ,
العدد 2
, محرم 1430 - كانون الثاني (يناير) 2009 |
| |
| The Cellular Mechanism of Insulin Resistance |
| آلية مقاومة الخلايا للأنسولين |
| Zaher Raslan |
| د. زاهر رسلان |
| الملخص Abstract |
| Diabetes mellitus is a metabolic disease that is characterized by an absolute or relative insulin deficiency or the resistance to insulin action by the insulin-responsive cells. Type2 diabetes is more predominant than type1 diabetes mellitus as it forms about 90 % of the diagnosed cases. Too much effort is being done at the moment to address the etiology of type2 diabetes mellitus, not only because it is more common but also because of the secondary complications that are associated with it, such as the cardiovascular disease (CVD), neuropathy, retinopathy and nephropathy. Insulin resistance plays an important role in the onset of type 2 diabetes mellitus yet the mechanisms responsible for this defect are still poorly understood. However, the recruitment of some advanced techniques such as the nuclear magnetic resonance spectroscopy (NMR) allowed us to improve our understanding about the onset of insulin resistance. In this review we address the latest breakthroughs in explaining the mechanism by which insulin resistance occurs such as the role of fatty acids and inflammatory response in it. |
| الداء السكري هو مرض استقلابي يتميز بعوز أنسولين مطلق أو نسبي، و يمكن أن ينجم عن مقاومة الخلايا للأنسولين من قبل الخلايا المستجيبة له. النمط الثاني للداء السكري هو أكثر انتشاراً من النمط الأول حيث أنه يشكل 90% من الحالات المشخصة. تبذل حالياً الكثير من الجهود لتوضيح سبب ظهور النمط الثاني من مرض السكري، ليس فقط لأنه أكثر شيوعاً من النمط الأول، بل بسبب المضاعفات الثانوية الخطيرة الناجمة عنه كالأمراض الوعائية القلبية (CVD)، الاعتلالات العصبية، الاعتلالات الشبكية بالإضافة إلى الاعتلالات الكلوية. مقاومة الأنسولين تلعب دوراً مهماً في ظهور النمط الثاني من مرض السكري ولكن الآلية التي تنجم عنها مقاومة الخلايا للأنسولين لا تزال غير مفهومة تماماً.
لقد سمحت الاستفادة من بعض التقنيات الحديثة كالرنين المغناطيسي النووي (NMR) بتطوير فهمنا حول كيفية نشوء مقاومة الخلايا للأنسولين. في هذا المقال سنحاول استعراض أهم المستجدات المتعلقة بفهم الآلية التي من خلالها تنشأ مقاومة الخلايا للأنسولين ومنها: دور الأحماض الدسمة والاستجابة الالتهابية في هذا الاعتلال.
|
| Introduction |
Diabetes Mellitus is the absolute or the relative insulin deficiency, or resistance to insulin action. It is a serious disease, which poses a major threat to people's life and has various complications as it may lead to the development of cardiovascular diseases, neuropathy, retinopathy, gastrointestinal diseases and other diseases. The percentage of people suffering from diabetes is increasing gradually year after year. In relation to the age of onset, etiological factors and the treatment scheme, diabetes mellitus can be divided into two types; Type1 diabetes mellitus or the insulin dependent diabetes mellitus (IDDM) and type2 diabetes or the non-insulin dependent diabetes mellitus (NIDDM). In type2 diabetes, tissues have a resistance toward insulin and/or abnormal insulin secretion. It has a mature onset and it forms 90% of the diagnosed cases. Insulin resistance is the major etiological factor in type 2 diabetes. As a result the full understanding of the mechanism by which insulin resistance occurs can give us a huge step forward in treating one of the major threats to man kind nowadays and also to predict prospective incidences of diabetes before they happen.
Insulin resistance can be defined as a reduced responsiveness to normal circulating levels of insulin (Savage et al., 2005). A reasonably good work has been done so far to establish the mechanism by which insulin resistance happens. In addition, in the last decade a breakthrough came up to link insulin resistance with the abnormal accumulation of fatty acids. This breakthrough basically diverted the focus on the glucose metabolism as a major reason for insulin resistance to another reason which is obesity. Not only do obese and diabetic people suffer from insulin resistance but also their offspring who are not diabetic (Rothman et al; 1995).
Basically, those findings were the outcome of using a technique called nuclear magnetic resonance spectroscopy (NMR).
|
| The pathogenesis of insulin resistance in skeletal muscles: |
| Using C13 NMR, it was confirmed that skeletal muscles are the major positions where insulin-stimulated glycogen synthesis takes place in both type 2 diabetics and in age- and weight-matched controls. In addition, it was found that in type 2 diabetics, patients suffer from a 50% less glycogen synthesis in skeletal muscles due to insulin resistance (Shulman et al., 1990). Three cellular enzymes were nominated as being the rate controlling step in the glycogen synthesis pathway. Those enzymes are hexokinaseII, glycogen synthase and glucose transport-4 GLUT-4. Their positions and their functions are illustrated in figure-1 (Shulman, 2000).
After that, the three possibilities
were reduced down into two. The two possible rate controlling steps became only hexokinase II and GLUT-4. This was confirmed after observing a significant reduction in glucose-6-phosphat increment. This deducted glycogen synthase from being the rate controlling step. This was applicable on both diabetic people (Rothman et al., 1992) and their offspring (Rothman et al., 1995), which confirmed more the fact that insulin resistance precedes 10-20 years the development of diabetes. Glucose transport (GLUT-4) was confirmed to be the rate controlling step when the levels of glucose were measured during a hyperinsulinemic and hyperglycemic clamps. The levels of glucose were way too less than it would have been if hexokinase II was the rate controlling step (Cline et al., 1999).
|
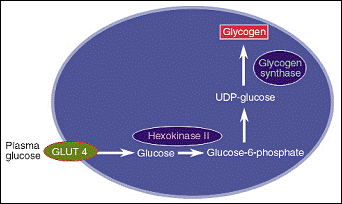
Figure 1: glycogen synthesis pathway and the role of hexokinase II, glycogen synthase and GLUT-4 (Shulman, 2000).
|
| The role of fatty acids in insulin resistance: |
It was Randle who first suggested that elevated free fatty acids in the plasma compete with glucose in cardiac muscle and leads to a fall in the percentage of glucose uptake. In addition, Randle mentioned the possibility of fatty acids contributing to insulin resistance (Randle et al., 1963). However he suggested a mechanism, illustrated in figure 2 (Shulman, 2000), which has been contradicted recently.
The mechanism by which FFA reduces glucose oxidation had been explained by using NMR technique that had been applied on healthy people under hyperindulinemic and euglycamic clamp conditions. The levels of glucose uptake as well as the levels of glycogen synthesis decreased after approximately 3.5 hours to approximately 40% and 50% respectively as a result of the infusion of elevated levels of fatty acids.
What is more, levels of glucose-6- phosphate significantly dropped
(Roden et al., 1996). Almost an identical protocol had been applied after that in addition to the measurement of intramuscular glucose levels and the IRS-1-associated IP3K activity. The outcome was that a defect in glucose transportation linked to an impaired IRS-1-associated IP3K activity is the reason behind the reduction of glucose uptake after an elevated FFA infusion (Dresner et al., 1999). The molecular mechanism by which IRS-1-associted PI3k activity is decreased has been established. An increase in plasma FFA leads to an augmented levels of intracellular acyl-CoA and diacylglycerol, which in turn leads to the activation of PKC-theta. The activation of PKC-theta results in a serin phosphorylation of IRS-1 instead of tyrosin phosphorylation. The outcome of serin phosphorylation would be the deactivation of IRS-1-associated PI3K. Subsequently, GLUT-4 is impaired (Yu et al., 2002). This mechanism is illustrated in figure (3) (Shulman, 2000).
Another study demonstrated a similar mechanism that induces insulin resistance in liver. When metabolite of lipids such as DAG and acyl CoA accumulate in hepatic cells they activate PKC-∑ which in it turn phosphorylates serin residues in the insulin receptor. This leads to a decreased activity of the receptor and decreased levels of tyrosin phosphorylation of IRS-2, which result in the deactivation of IRS-2-stimulated IP3k (Samuel et al., 2004).
Elevated FFA is not only restricted to type2 diabetes, but it is also spotted in all forms of insulin resistance states such as obesity and offspring of type2 diabetics. The molecular mechanism behind FFA ectopic accumulation can be explained by the hypothesis that addresses a role of genetic defects that affect mitochondrial biogenesis. Those genetic alterations are thought to include mutations in enzymes such as peroxisome proliferators-activated receptor gamma-coactivator-1α (PGC-1α), AMP kinase and Ca+2/calmodulin-dependent protein kinase IV (CAMKIV) (Savage et al., 2005).
|
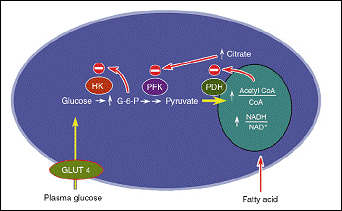
Figure 2: The mechanism by which fatty acids induces insulin as suggested by Randle (Shulman, 2000).
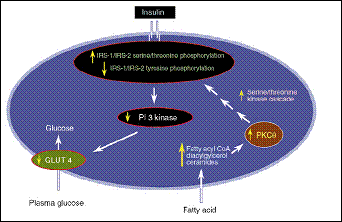
Figure 3: The mechanism by which FFA induces insulin resistance
|
| The role of inflammatory response in insulin resistance: |
| Not only does obesity induce insulin resistance but also it induces several inflammatory responses that are characterized by an increase in infiltration of macrophages that are responsible for inflammatory response (Wellen and Hotamisligil, 2003). This has been linked afterward to the induction of insulin resistance based on the fact that, FFA ectopic accumulation and inflammatory response take place in the same insulin sensitive tissues; hepatic tissue, adipose tissues and skeletal muscle. In the light of that, cytokines such as TNF-α and IL-6 have been linked to insulin resistance. Consistently, the inactivation of TNF-α led to an improvement in insulin sensitivity (Hotamisligil, 2003). Other cytokines have been linked to induce insulin resistance by phosphorylating serin residues at different sites in IRS-1. C-jun N-terminal Kinase (JNK) is one of those cytokines, which is confirmed to phosphorylate IRS-1 at serin 307 in vitro, and it is thought now that it could be a major factor in inducing insulin resistance and type 2 diabetes (Hotamisligil, 2003). In addition, IkappaB kinase beta (IKKbeta) has also been linked to insulin resistance as the overexpression of this inflammatory signaling mediator led to induce insulin resistance, whereas the salicylate-mediated inhibition of it and the engineering of heterozygote IKKbeta+/- mice led to increase insulin sensitivity (Yuan et al., 2001). In addition, it was found that suppressor of cytokine signaling proteins (SOCS-3) also has a linking role in insulin resistance. SOCS-3 signaling proteins function as a negative regulator for cytokines signaling and they are highly expressed in the liver and muscles in obese rodents. Adenovirus-mediated transfection of SOCS-3 to a liver, led to inhibit the tyrosin phosphorylation of insulin receptor. It is thought that SOCS-3 participates in insulin resistance through a direct inhibition of insulin receptor by binding to tyr960 at which IRS-1 is recognized. This is confirmed in vitro (Ueki et al., 2004). What is more, SOCS-3 was found to induce leptin resistance in leptin responsive tissue which leads to an increase in body fat (Bjorbaek et al., 1999). Another uncertain factor that may play a role in insulin resistance is cytokine-like proteins or adipokines, which are either produced by the adipose tissue or by the macrophages (Savage et al., 2005). A hypothesis suggests a role of macrophage infiltration to white adipose tissue in mice and a consequent upregulation of inflammatory genes. This was followed by augmented levels of circulating insulin and was contrasted by insulin sensitizers (Xu et al., 2003). This is not a strong hypothesis and the molecular mechanism is still to be elucidated. Finally a relationship between plasma elevated levels of adiponectin and insulin sensitivity has been suggested, but a lot of questions are still unanswered regarding the mechanism by which it exerts its function and the impact on insulin resistance (Berg et al., 2002). |
| Conclusion: |
|
Using NMR technique, we now have a reasonable insight about the mechanism by which insulin resistance occurs. In addition, the rate controlling step for fat-stimulated insulin resistance has been identified. Defects in glucose transport are behind the insulin resistance-mediated decrease in glucose uptake in insulin-sensitive tissues. In addition, a role for the ectopic accumulation of FFA in inducing defects in GLUT-4 has been explained. However, there are still a lot of questions to be answered in respect to the involvement of cytokines and the role of inflammatory response, triggered by the accumulation of FFA, in mediating insulin resistance and they are likely to be secondary factors in triggering insulin resistance. As a result, more studies should be done to get a perfect understanding about insulin resistance as it can produce a significant improvement in treating type2 diabetes and the metabolic syndrome.
|
| References: |
1-Berg A.H; Combs T.P. and Scherer P.E.
ACRP30 adiponectin: an adipokine regulating glucose and lipid metabolism.
Trends Endocrinol Metab; 13, 84-89, 2002.
2-Bjorbaek C; El-Haschimi K; Frantz, J.D. and Flier J.S.
The role of SOCS-3 in leptin signaling and leptin resistance.
J Biol Chem; 274, 30059-30065, 1999.
3-Cline G.W; Petersen K.F; Krssak M; Shen J; Hundal R.S; Trajanoski Z; Inzucchi S; Dresner A; Rothman D.L. and Shulman G.I.
Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes.
N Engl J Med; 341, 240-246, 1999.
4-Dresner A; Laurent D; Marcucci M; Griffin M.E; Dufour S; Cline G. W; Slezak L.A; Andersen D.K; Hundal R.S. and Rothman D.L.
Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity.
J Clin Invest, 103, 253-259, 1999.
5-Hotamisligil, G. S.
Inflammatory pathways and insulin action.
Int J Obes Relat Metab Disord; 27 Suppl 3, S53-55, 2003.
6-Randle P.J; Garland P.B; Hales C.N. and Newsholme E.A.
The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus.
Lancet, 1, 785-789, 1963.
7-Roden M; Price T.B; Perseghin G; Petersen K.F; Rothman D.L; Cline G.W; and Shulman G.I.
Mechanism of free fatty acid-induced insulin resistance in humans.
J Clin Invest, 97, 2859-2865, 1996.
8-Rothman D.L; Magnusson, I; Cline, G; Gerard, D; Kahn, C. R; Shulman, R.G. and Shulman, G.I.
Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus.
Proc Natl Acad Sci U S A; 92, 983-987, 1995.
9-Rothman D.L; Shulman R.G. and Shulman G.I.
31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus.
J Clin Invest, 89, 1069-1075, 1992.
10-Samuel V.T; Liu Z.X; Qu X; Elder B.D; Bilz S; Befroy D; Romanelli A.J. and Shulman G.I.
Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease.
J Biol Chem, 279, 32345-32353, 2004.
11-Savage D.B; Petersen K.F. and Shulman G.I.
Mechanisms of insulin resistance in humans and possible links with inflammation.
Hypertension, 45, 828-833, 2005.
12-Shulman G.I.
Cellular mechanisms of insulin resistance.
J Clin Invest, 106, 171-176, 2000.
13-Shulman G.I; Rothman D.L; Jue T; Stein P; DeFronzo R.A. and Shulman R.G.
Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy.
N Engl J Med; 322, 223-228, 1990.
14-Ueki K; Kondo T. and Kahn C.R.
Suppressor of cytokine signaling 1 (SOCS-1) and SOCS-3 cause insulin resistance through inhibition of tyrosine phospho-rylation of insulin receptor substrate proteins by discrete mechanisms.
Mol Cell Biol; 24, 5434-5446, 2004.
15-Wellen K.E. and Hotamisligil G.S.
Obesity-induced inflammatory changes in adipose tissue.
J Clin Invest, 112, 1785-1788, 2003.
16-Xu H; Barnes G.T; Yang Q; Tan G; Yang D; Chou C.J; Sole J; Nichols A; Ross J.S; Tartaglia L.A. and Chen, H.
Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance.
J Clin Invest, 112, 1821-1830, 2003.
17-Yu C; Chen Y; Cline G.W; Zhang D; Zong H; Wang Y; Bergeron R; Kim J.K; Cushman S. W. and Cooney G.J.
Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle.
J Biol Chem; 277, 50230-50236, 2002.
18-Yuan M; Konstantopoulos N; Lee J; Hansen L; Li Z.W; Karin M; and Shoelson S.E.
Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta.
Science, 293, 1673-1677, 2001.
|
| |
| المجلد 5 ,
العدد 2
, محرم 1430 - كانون الثاني (يناير) 2009 |
|
|
|
