1. Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, et al. Heart
disease and stroke statistics-2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009;119:e21–181.
2. Asplund K, Stegmayr B, Peltonen M. From the twentieth to the twenty-first century: a public
health perspective on stroke. In: Ginsberg MD, Bogousslavsky J, eds. Cerebrovascular disease pathophysiology, diagnosis, and management. Vol. 2. Malden (MA): Blackwell Science; 1998.
3. Adams HPJ, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE 3rd. Classification of subtype of acute ischemic stroke: definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment Stroke 1993;24:35– 41.
4. Easton JD, Saver JL, Albers GW, Alberts MJ, Chaturvedi S, Feldmann E, et al. Definition and evaluation of transient ischemic attack: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association Stroke Council; Council on
Cardiovascular Surgery and Anesthesia; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; and the Interdisciplinary Council on Peripheral Vascular Disease: the American Academy of Neurology affirms the value of this statement as an educational tool for neurologists. Stroke 2009;40: 2276–93.
5. Albers GW, Caplan LR, Easton JD, Fayad PB, Mohr JP, Saver JL, et al. Transient ischemic
Attack-proposal for a new definition. N Engl J Med 2002;347:1713–6.
6. Levy DE. How transient are transient ischemic attacks? Neurology 1988;38:674–7.
7. Adams HP Jr, Del Zoppo GJ, Alberts MJ, Bhatt DL, Brass L, Furlan A, et al. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart
Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups: the American Academy of Neurology affirms the value of this guideline as an educational tool for neurologists. Circulation 2007;115:e478–534.
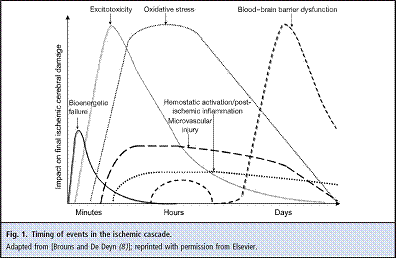
8. Brouns R, De Deyn PP. The complexity of neurobiological processes in acute ischemic stroke. Clin Neurol Neurosurg 2009;111:483–95.
9. Testai FD, Aiyagari V. Acute hemorrhagic stroke pathophysiology and medical interventions: blood pressure control, management of anticoagulant-associated brain hemorrhage and general management principles. Neurol Clin 2008;26:963–85.
10. Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol 2006;5:53–63.
11. Morgenstern LB, Lisabeth LD, Mecozzi AC, Smith MA, Longwell PJ, McFarling DA, Risser
JM. A population-based study of acute stroke and TIA diagnosis. Neurology 2004;62:895-900.
12. Hand PJ, Kwan J, Lindley RI, Dennis MS, Wardlaw JM. Distinguishing between stroke and mimic at the bedside: the brain attack study. Stroke 2006;37:769–75.
13. Goldstein LB, Samsa GP. Reliability of the National Institutes of Health Stroke Scale: extension to non-neurologists in the context of a clinical trial. Stroke 1997;28:307–10.
14. Tan JC, Dillon WP, Liu S, Adler F, Smith WS, Wintermark M. Systematic comparison of
perfusion-CT and CT-angiography in acute stroke patients. Ann Neurol 2007;61:533– 43.
15. Kohrmann M, Schellinger PD. Acute stroke triage to intravenous thrombolysis and other therapies with advanced CT or MR imaging: pro MR imaging. Radiology 2009;251:627–33.
16. Gonzalez RG, Schaefer PW, Buonanno FS, Schwamm LH, Budzik RF, Rordorf G, et al.
Diffusion-weighted MR imaging: diagnostic accuracy in patients imaged within 6 hours of
stroke symptom onset. Radiology 1999;210: 155–62.
17. Schaefer PW, Grant PE, Gonzalez RG. Diffusionweighted MR imaging of the brain. Radiology 2000;217:331–45.
18. Schulz UG, Briley D, Meagher T, Molyneux A, Rothwell PM. Diffusion-weighted MRI in 300 patients presenting late with subacute transient ischemic attack or minor stroke. Stroke 2004; 35:2459–65.
19. Kang DW, Latour LL, Chalela JA, Dambrosia J, Warach S. Early ischemic lesion recurrence within a week after acute ischemic stroke. Ann Neurol 2003;54:66–74.
20. Wen HM, Lam WW, Rainer T, Fan YH, Leung TW, Chan YL, Wong KS. Multiple acute cerebral infarcts on diffusion-weighted imaging and risk of recurrent stroke.
Neurology 2004;63:1317–9.
21. Coutts SB, Hill MD, Simon JE, Sohn CH, Scott JN, Demchuk AM. Silent ischemia in minor stroke and TIA patients identified on MR imaging. Neurology 2005;65:513–7.
22. Sylaja PN, Coutts SB, Subramaniam S, Hill MD, Eliasziw M, Demchuk AM. Acute ischemic lesions of varying ages predict risk of ischemic events in stroke/TIA patients. Neurology 2007; 68:415–9.
23. Ringelstein EB. Ultrafast magnetic resonance imaging protocols in stroke. J Neurol Neurosurg Psychiatry 2005;76:905.
24. U-King-Im JM, Trivedi RA, Graves MJ, Harkness K, Eales H, Joubert I, et al. Utility of an ultrafast magnetic resonance imaging protocol in recent and semi-recent strokes. J Neurol Neurosurg Psychiatry 2005;76:1002–5.
25. Grotta JC, Chiu D, Lu M, Patel S, Levine SR, Tilley BC, et al. Agreement and variability in the interpretation of early CT changes in stroke patients qualifying for intravenous rtPA therapy. Stroke 1999;30:1528-33.
26. Kalafut M, Schriger DL, Saver JL, Starkman S. Detection of early CT signs of -1/3 middle
cerebral artery infarctions: interrater reliability and sensitivity of CT interpretation by physicians involved in acute stroke care. Stroke 2000;31: 1667–71.
27. Schriger DL, Kalafut M, Starkman S, Krueger M, Saver JL. Cranial computed tomography interpretation in acute stroke: physician accuracy in determining eligibility for thrombolytic therapy. JAMA 1998;279:1293–7.
28. Hacke W, Kaste M, Bluhmki E, Brozman M, Da´ valos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 2008;359:1317–29.
29. Katzan IL, Hammer MD, Hixson ED, Furlan AJ, Abou-Chebl A, Nadzam DM. Utilization of intravenous tissue plasminogen activator for acute ischemic stroke.
Arch Neurol 2004;61:346–50.
30. Wang DZ, Rose JA, Honings DS, Garwacki DJ, Milbrandt JC. Treating acute stroke patients with intravenous tPA. The OSF Stroke Network experience.
Stroke 2000;56:1015–20.
31. Kleindorfer D, Kissela B, Schneider A, Woo D, Khoury J, Miller R, et al. Eligibility for recombinant tissue plasminogen activator in acute ischemic stroke: a population-based study. Stroke 2004;35:e27–9.
32. Davidson MH, Corson MA, Alberts MJ, Anderson JL, Gorelick PB, Jones PH, et al. Consensus panel recommendation for incorporating lipoprotein-associated phospholipase A2 testing into cardiovascular disease risk assessment guidelines.
Am J Cardiol 2008;101:51F–57F.
33. Gorelick PB. Lipoprotein-associated phospholipase A2 and risk of stroke. Am J Cardiol 2008;101:34F–40F
34. Elkind MS, Tai W, Coates K, Paik MC, Sacco RL. Lipoprotein-associated phospholipase A2 activity and risk of recurrent stroke. Cerebrovasc Dis 2009;27:42–50.
35. Oei H-H, van der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MMB, Witteman JCM. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation 2005;111:570–5.
36. Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR, Chambless LE, et al. Lipoproteinassociated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Arch Intern Med 2005;165:2479–84.
37. Wassertheil-Smoller S, Hendrix SL, Limacher M, Heiss G, Kooperberg C, Baird A, et al. Effect of estrogen plus progestin on stroke in postmenopausal women: the Women’s Health Initiative: a randomized trial. JAMA 2003;289:2673–84.
38. Wassertheil-Smoller S, Kooperberg C, McGinn AP, Kaplan RC, Hsia J, Hendrix SL, et al.
Lipoprotein-associated phospholipase A2, hormone use, and the risk of ischemic stroke in
postmenopausal women. Hypertension 2007; 51:1115–22.
39. McConnell JP, Jaffe AS. Variability of lipoprotein-associated phospholipase A2 measurements. Clin Chem 2008;54:932–3.
40. Achan V, Broadhead M, Malaki M, Whitley G, Leiper J, MacAllister R, Vallance P. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol 2003;23: 1455–9.
41. Dayoub H, Achan V, Adimoolam S, Jacobi J, Stuehlinger MC, Wang BY, et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation 2003;108:3042–7.
42. Kielstein JT, Bode-Boger SM, Frolich JC, Ritz E, Haller H, Fliser D. Asymmetric dimethylargininearginine, blood pressure, and renal perfusion in elderly subjects. Circulation 2003;107:1891–5.
43. Miyazaki H, Matsuoka H, Cooke JP, Usui M, Ueda S, Okuda S, Imaizumi T. Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis. Circulation 1999;99:1141–6.
44. Masuda H, Goto M, Tamaoki S, Azuma H. Accelerated intimal hyperplasia and increased endogenous inhibitors for NO synthesis in rabbits with alloxan-induced hyperglycaemia. Br J Pharmacol 1999;126:211–8.
45. Stuhlinger MC, Oka RK, Graf EE, Schmolzer I, Upson BM, Kapoor O, et al. Endothelial dysfunction induced by hyperhomocysteinemia: role of asymmetric dimethylarginine. Circulation 2003;108:933–8.
46. Boger RH, Lentz SR, Bode-Boger SM, Knapp HR,Haynes WG. Elevation of asymmetrical dimethylarginine may mediate endothelial dysfunction during experimental hyperhomocysteinemia in humans. Clin Sci (Lond) 2001;100:161–7.
47. Boger RH, Bode-Boger SM, Sydow K, Heistad DD, Lentz SR. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in
monkeys with hyperhomocyst(e)inemia or hypercholesterolemia. Arterioscler Thromb Vasc
Biol 2000;20:1557–64.
48. Zoccali C, Mallamaci F, Maas R, Benedetto FA, Tripepi G, Malatino LS, et al. Left ventricular hypertrophy, cardiac remodeling and asymmetric dimethylarginine (ADMA) in hemodialysis patients. Kidney Int 2002;62:339–45.
49. Bode-Boger SM, Boger RH, Kienke S, Junker W, Frolich JC. Elevated L arginine/dimethylarginine ratio contributes to enhanced systemic NO production by dietary L-arginine in hypercholesterolemic rabbits. Biochem Biophys Res Commun1996;219:598–603.
50. Yu X, Li Y, Xiong Y. Increase of an endogenous inhibitor of nitric oxide synthesis in serum of high cholesterol fed rabbits. Life Sci 1994;54: 753–8.
51. Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 1998;98:1842–7.
52. Yoo J-H, Lee S-C. Elevated levels of plasma homocyst(e)ine and asymmetric dimethylarginine in elderly patients with stroke. Atherosclerosis 2001;158:425–30.
53. Leong T, Zylberstein D, Graham I, Lisner L, Ward D, Fogarty J, et al. Asymmetric dimethylarginine independently predicts fatal and nonfatal myocardial infarction and stroke in women: 24-year follow-up of the population study of women in Gothenberg. Arterioscler Thromb Vasc Biol 2008;28:961–7.
54. Pikula A, Boger RH, Beiser AS, Maas R, DeCarli C, Schwedhelm E, et al. Association of plasma ADMA levels with MRI markers of vascular brain injury. Stroke 2009;40:2959–64.
55. Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004;4:617–29.
56. Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci 2005;6:931–44.
57. Clark AW, Krekoski CA, Bou S-S, Chapman KR, Edwards DR. Increased gelatinase A (MMP-2) and gelatinase B (MMP-9) activities in human brain after focal ischemia. Neurosci Lett 1997;238:53–6.
58. Anthony DC, Ferguson B, Matyzak MK, Miler KM, Esiri MM, Perry VH. Differential matrix metalloproteinases expression in cases of multiple sclerosis and stroke. Neuropathol Appl Neurobiol 1997;23:406–15.
59. Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res 2002;69:1–9.
60. Rosenberg GA, Navrati LM, Barone F, Feuerstein G. Proteolytic cascade enzymes increase in focal cerebral ischemia in rat. J Cereb Blood Flow Metab 1996;16:360–6.
61. del Zoppo GJ. Stroke and neurovascular protection. N Engl J Med 2006;354:553–5.
62. Lo EH, Broderick JP, Moskowitz MA. tPA and proteolysis in the neurovascular unit.
Stroke 2004;35:354–6.
63. Montaner J, Alvarez-Sab?´n J, Molina C, Angle´ s A, Abilleira S, Arenillas J, et al. Matrix metalloproteinase expression after human cardioembolic stroke: temporal profile and relation to neurological impairment. Stroke 2001;32:1759–66.
64. Alvarez-Sab?´n J, Delgado P, Abilleira S, Molina CA, Arenillas J, Ribo´ M, et al. Temporal profile of matrix metalloproteinases and their inhibitors after spontaneous intracerebral hemorrhage: relationship to clinical and radiological outcome. Stroke 2004;35:1316–22.
65. Vukasovic I, Tesija-Kuna A, Topic E, Supanc V, Demarin V, Petrovcic M. Matrix metalloproteinases and their inhibitors in different acute stroke subtypes.
Clin Chem Lab Med 2006;44:428–34.
66. Montaner J, Alvarez-Sab?´n J, Molina CA, Angle´ s A, Abilleira S, Arenillas J, Monasterio J. Matrix metalloproteinase expression is related to hemorrhagic transformation after cardioembolic stroke. Stroke 2001;32:2762–7.
67. Montaner J, Molina CA, Monasterio J, Abilleira S, Arenillas JF, Ribo´ M, et al. Matrix metalloproteinase- 9 pretreatment level predicts intracranial hemorrhagic complications after thrombolysis in human stroke. Circulation 2003;107: 598–603.
68. Rosell A, Alvarez-Sab?´n J, Arenillas JF, Rovira A, Delgado P, Ferna´ ndez-Cadenas I, et al. A matrix metalloproteinase protein array reveals a strong relation between MMP-9 and MMP-13 with diffusion-weighted image lesion increase in human stroke.
Stroke 2005;36:1415–20.
69. Horstmann S, Kalb P, Koziol J, Gardner H, Wagner S. Profiles of matrix metalloproteinases and their inhibitors, and laminin in stroke patients: influence of different therapies. Stroke 2003;34: 2165–70.
70. Ning M, Furie KL, Koroshetz WJ, Lee H, Barron M, Lederer M, et al. Association between tPA therapy and raised early matrix metalloproteinase-9 in acute stroke.
Neurology 2006; 66:1550–5.
71. Moore BW. A soluble protein characteristic of the nervous system. Biochem Biophys Res Commun 1965;19:739–44.
72. Donato R. S100: a multigenic family of calciummodulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol 2001;33:637– 68.
73. Kapural M, Krizanac-Bengez L, Barnett G, Perl J, Masaryk T, Apollo D, et al. Serum S-100beta as a possible marker of blood–brain barrier disruption.Brain Res 2002;940:102– 4.
74. Kanner AA, Marchi N, Fazio V, Mayberg MR, Koltz MT, Siomin V, et al. Serum S100beta: a noninvasive marker of blood-brain barrier function and brain lesions.
Cancer 2003;97:2806–13.
75. Persson L, Hardemark HG, Gustafsson J, Rundstro ¨m G, Mendel-Hartvig I, Esscher T, P?hlman S. S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke 1987;18:911–8.
76. Abraha HD, Butterworth RJ, Bath PM, Wassif WS, Garthwaite J, Sherwood RA. Serum S-100 protein, relationship to clinical outcome in acute stroke.
Ann Clin Biochem 1997;34:366–70.
77. Buttner T, Weyers S, Postert T, Sprengelmeyer R, Kuhn W. S-100 protein: serum marker of focal brain damage after ischemic territorial MCA infarction. Stroke 1997;28:1961–5.
78. Elting JW, de Jager AE, Teelken AW, Schaaf MJ, Maurits NM, van der Naalt J, et al. Comparison of serum S-100 protein levels following stroke and traumatic brain injury. J Neurol Sci 2000; 181:104–10.
79. Wunderlich MT, Wallesch CW, Goertler M. Release of neurobiochemical markers if brain damage is related to the neurovascular status on admission and the site of arterial occlusion in acute ischemic stroke. J Neurol Sci 2004;227:49–53.
80. Jauch EC, Lindsell C, Broderick J, Fagan SC, Tilley BC, Levine SR; NINDS rt-PA Stroke Study Group. Association of serial biochemical markers with acute ischemic stroke: the National Institute of Neurological Disorders and Stroke recombinant tissue plasminogen activator Stroke Study. Stroke 2006;37:2508–13.
81. Foerch C, Du Mesnil de Rochemont R, Singer O, Neumann-Haefelin T, Buchkremer M, Zanella FE, et al. S100B as a surrogate marker for successful clot lysis in hyperacute middle cerebral artery occlusion. J Neurol Neurosurg Psychiatry 2003;74:322–5.
82. Foerch C, Singer OC, Neumann-Haefelin T, du Mesnil de Rochemont R, Steinmetz H, Sitzer M. Evaluation of serum S100B as a surrogate marker for long-term outcome and infarct volume in acute middle cerebral artery infarction. Arch Neurol 2005;62:1130–4.
83. Jonsson H, Johnsson P, Birch-Iensen M, Alling C, Westaby S, Blomquist S. S100B as a predictor of size and outcome of stroke after cardiac surgery.
Ann Thorac Surg 2001;71:1433–7.
84. Hill MD, Jackowski G, Bayer N, Lawrence M, Jaeschke R. Biochemical markers in acute ischemic stroke. CMAJ 2000;162:1139–40.
85. Ishiguro Y, Kato K, Ito T, Nagaya M. Determination of three enolase isozymes and S-100
protein in various tumors in children. Cancer Res 1983;43:6080–4.
86. Raabe A, Grolms C, Keller M, Do¨ hnert J, Sorge O, Seifert V. Correlation of computed tomography findings and serum brain damage markers following severe head injury. Acta Neurochir 1998;140:791–2.
87. Dambinova SA, Khounteev GA, Izykenova GA, Zavolokov IG, Ilyukhina AY, Skoromets AA. Blood test detecting autoantibodies to N-methyl- D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke.Clin Chem 2003;49:1752–62.
88. Bokesch PM, Izykenova GA, Justice JB, Easley KA, Dambinova SA. NMDA receptor antibodies predict adverse neurological outcome after cardiac surgery in high-risk patients. Stroke 2006; 37:1432–6.
89. Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP thirty-one years (1969–
2000). Neurochem Res 2000;25:1439–51.
90. Niebro´ j-Dobosz I, Rafa?owska J, Lukasiuk M, Pfeffer A, Mossakowski MJ. Immunochemical analysis of some proteins in cerebrospinal fluid and serum of patients with ischemic strokes. Folia Neuropathol 1994;32:129–37.
91. Herrmann M, Vos P, Wunderlich MT, de Bruijn CH, Lamers KJ. Release of glial tissue-specific proteins after acute stroke: a comparative analysis of serum concentrations of protein S-100B and glial fibrillary acidic protein. Stroke 2000; 31:2670–7.
92. Foerch C, Singer O, Neumann-Haefelin T, Raabe A, Sitzer M. Utility of serum GFAP in monitoring acute MCA territorial infarction. Cerebrovasc Dis 2003;16:45.
93. Foerch C, Curdt I, Yan B, Dvorak F, Hermans M, Berkefeld J, et al. Serum glial fibrillary acidic protein as a biomarker for intracerebral haemorrhage in patients with acute stroke. J Neurol Neurosurg Psychiatry 2006;77:181–4.
94. Dvorak F, Haberer I, Sitzer M, Foerch C. Characterisation of the diagnostic window of serum glial fibrillary acidic protein for the differentiation of intracerebral haemorrhage and ischaemic stroke. Cerebrovasc Dis 2009;27:37– 41.
95. Unden J, Strandberg K, Malm J, Campbell E, Rosengren L, Stenflo J, et al. Explorative investigation of biomarkers of brain damage and coagulation system activation in clinical stroke differentiation. J Neurol 2009;256:72–7.
96. Nagakubo D, Taira T, Kitaura H, Ikeda M, TamaiK, Iguchi-Ariga SM, Ariga H. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun 1997;231:509–13.
97. Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science (Wash DC) 2003;299:256–9.
98. Lescuyer P, Allard L, Zimmermann-Ivol CG, Burgess JA, Hughes-Frutiger S, Burkhard PR, et al. Identification of post-mortem cerebrospinal fluid proteins as potential biomarkers of ischemia and neurodegeneration. Proteomics 2004; 4:2234–41.
99. Allard L, Burkhard PR, Lescuyer P, Burgess JA, Walter N, Hochstrasser DF, Sanchez JC. PARK7 and nucleoside diphosphate kinase A as plasma markers for the early diagnosis of stroke. Clin Chem 2005;51:2043–51.
100. Reynolds MA, Kirchick HJ, Dahlen JR, Anderberg JM, McPherson PH, Nakamura KK, et al. Early biomarkers of stroke. Clin Chem 2003;49:1733–9.
101. Lynch JR, Blessing R, White WD, Grocott HP, Newman MF, Laskowitz DT. Novel diagnostic test for acute stroke. Stroke 2004;35:57–63.
102. Laskowitz DT, Blessing R, Floyd J, White WD, Lynch JR. Panel of biomarkers predicts stroke. Ann N Y Acad Sci 2005;1053:30.
103. Laskowitz DT, Kasner SE, Saver J, Remmel KS, Jauch EC, Group BS. Clinical usefulness of a biomarker-based diagnostic test for acute stroke: the biomarker rapid assessment in ischemic injury (BRAIN) study. Stroke 2009; 40:77–85.
104. Whiteley W, Tseng M-C, Sandercock P. Blood biomarkers in the diagnosis of ischemic stroke: a systematic review. Stroke 2008; 39: 2902–9.
105. Whiteley W, Chong WL, Sengupta A, Sandercock P. Blood markers for the prognosis of
ischemic stroke: a systematic review. Stroke 2009;40:e380–9.
106. Apple FS, Chung AY, Kogut ME, Bubany S, Murakami MM. Decreased patient charges following implementation of point-of-care cardiac troponin monitoring in acute coronary syndrome patients in a community hospital cardiology unit. Clin Chem Acta 2006;370:191–5.
107. Singer AJ, Ardise J, Gulla J, Cangro J. Point-ofcare testing reduces length of stay in emergency department chest pain patients. Ann Emerg Med 2005;45:587–91.
108. Ryan RJ, Lindsell CJ, Hollander JE, O’Neil B, Jackson R, Schreiber D, et al. A multicenter randomized controlled trial comparing central laboratory and point-of-care cardiac marker testing strategies: the Disposition Impacted by Serial Point of Care Markers in Acute Coronary Syndromes (DISPO-ACS) trial. Ann Emerg Med 2008;53:321– 8.
109. Storrow AB, Lyon JA, Porter MW, Zhou C, Han JH, Lindsell CJ. A systematic review of emergency department point-of-care cardiac markers and efficiency measures.
Point Care 2009;8:121–5.
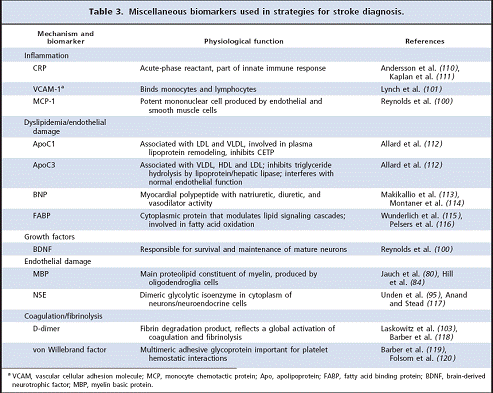
110. Andersson J, Johansson L, Ladenvall P, Wiklund P-G, Stegmayr B, Jern C, Bomana K. C-reactive protein is a determinant of first-ever stroke: prospective nested case-referent study. Cerebrovasc Dis 2009;27:544–51.
111. Kaplan RC, McGinn AP, Baird AE, Hendrix SL, Kooperberg C, Lynch J, et al. Inflammation and hemostasis biomarkers for predicting stroke in postmenopausal women: the Women’s Health Initiative Observational Study. J Stroke Cerebrovasc Dis 2008;17:344–55.
112. Allard L, Lescuyer P, Burgess J, Leung KY, Ward M, Walter N, et al. ApoC-I and ApoC-III as potential plasmatic markers to distinguish between ischemic and hemorrhagic stroke. Proteomics 2004;4:2242–51.
113. Makikallio AM, Makikallio TH, Korpelainen JT, Vuolteenaho O, Tapanainen JM, Ylitalo K, et al. Natriuretic peptides and mortality after stroke. Stroke 2005;36:1016–20.
114. Montaner J, Perea-Gainza M, Delgado P, Ribo´ M, Chaco´ n P, Rosell A, et al. Etiologic diagnosis of ischemic stroke subtypes with plasma biomarkers.Stroke 2008;39:2280–7.
115. Wunderlich MT, Hanhoff T, Goertler M, Spener F, Glatz JF, Wallesch CW, Pelsers MM. Release of brain-type and heart-type fatty acid-binding proteins in serum after acute ischaemic stroke. J Neurol 2005;252:718–24.
116. Pelsers MMAL, Hanhoff T, Van der Voort D, Arts B, Peters M, Ponds R, et al. Brain-type and heart-type fatty acid-binding proteins in the brain; tissue distribution and clinical utility. Clin Chem 2004;50:1568–75.
117. Anand N, Stead LG. Neuron-specific enolase as a marker for acute ischemic stroke: a systematic review. Cerebrovasc Dis 2005;20:213–9.
118. Barber M, Langhorne P, Rumley A, Lowe GD, Stott DJ. D-dimer predicts early clinical progression in ischemic stroke: confirmation using routine clinical assays.
Stroke 2006;37:1113–5.
119. Barber M, Langhorne P, Rumley A, Lowe GD, Stott DJ. Hemostatic function and progressing ischemic stroke: D-dimer predicts early clinical progression. Stroke 2004;35:1421–5.
120. Folsom AR, Rosamond WD, Shahar E, Cooper LS, Aleksic N, Nieto FJ, et al. Prospective study of markers of hemostatic function with risk of ischemic stroke. Circulation 1999;100:736–42
|